Getting started with InstaNovo¶
In this notebook, we demo InstaNovo, a transformer neural network with the ability to translate fragment ion peaks into the sequence of amino acids that make up the studied peptide(s). We evaluate the model on the yeast test fold of nine-species dataset.
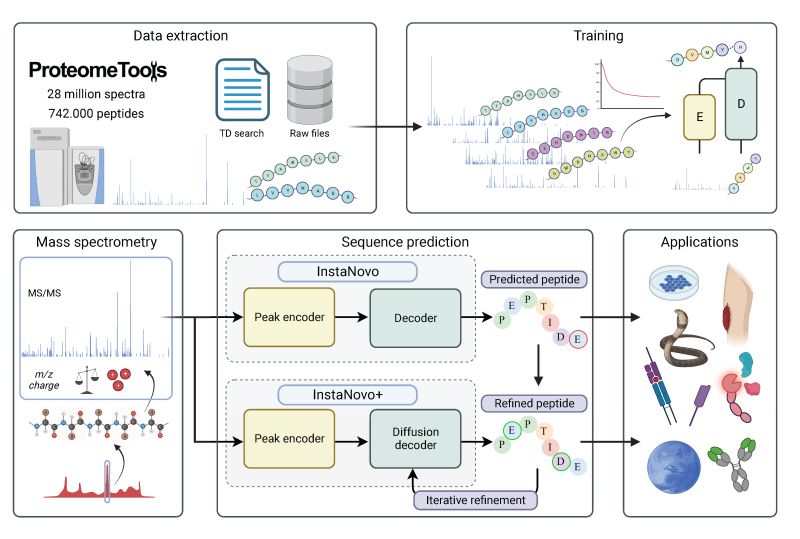
Links:
- Nature Machine Intelligence Paper: InstaNovo enables diffusion-powered de novo peptide sequencing in large-scale proteomics experiments
Kevin Eloff, Konstantinos Kalogeropoulos, Amandla Mabona, Oliver Morell, Rachel Catzel, Esperanza Rivera-de-Torre, Jakob Berg Jespersen, Wesley Williams, Sam P. B. van Beljouw, Marcin J. Skwark, Andreas Hougaard Laustsen, Stan J. J. Brouns, Anne Ljungars, Erwin M. Schoof, Jeroen Van Goey, Ulrich auf dem Keller, Karim Beguir, Nicolas Lopez Carranza, Timothy P. Jenkins - Code: GitHub
Important:
It is highly recommended to run this notebook in an environment with access to a GPU. If you are running this notebook in Google Colab:
- In the menu, go to
Runtime > Change Runtime Type > T4 GPU
Loading the InstaNovo model¶
We first install the latest instanovo from PyPi
try:
import instanovo
except ImportError:
!uv pip install "instanovo[cu126]>=1.2.2" pyopenms-viz
print('Installation complete. Restarting runtime to apply changes...')
import os
os.kill(os.getpid(), 9)
# Filter warnings and set logging level
import warnings
import logging
warnings.filterwarnings("ignore", module="matplotlib")
warnings.filterwarnings("ignore", module="torch")
logging.getLogger("matplotlib").setLevel(logging.WARNING)
logging.getLogger("rdkit").setLevel(logging.WARNING)
We can use instanovo version to check the version of InstaNovo (the transformer-based model), InstaNovo+ (the diffusion-based model) and some of their dependencies.
!instanovo version
Import the transformer-based InstaNovo model.
from instanovo.transformer.model import InstaNovo
Set the device to GPU if available (recommended), otherwise use CPU.
import torch
device = "cuda" if torch.cuda.is_available() else "cpu"
device
InstaNovo supports automatic model downloads. You can see the IDs of the pretrained models that are available.
InstaNovo.get_pretrained()
And download the model checkpoint given the ID.
model, config = InstaNovo.from_pretrained("instanovo-v1.2.0")
model = model.to(device).eval()
Alternatively, you can also download the model checkpoint directly from the InstaNovo releases page.
Loading the nine-species dataset¶
Download the yeast test fold of the nine-species dataset dataset from HuggingFace.
We can use our SpectrumDataFrame to download this directly. SpectrumDataFrame is a special data class used by InstaNovo to read and write from multiple formats, including mgf, mzml, mzxml, pandas, polars, HuggingFace, etc.
from instanovo.utils.data_handler import SpectrumDataFrame
sdf = SpectrumDataFrame.from_huggingface(
"InstaDeepAI/ms_ninespecies_benchmark",
is_annotated=True,
shuffle=False,
split="test[:1%]", # Let's only use a tiny subset of the test data for faster inference in this notebook
)
sdf.to_pandas().head(5)
Let's quickly plot the spectrum in the first row
import pandas as pd
pd.options.plotting.backend = "ms_matplotlib"
row = sdf[0]
row_df = pd.DataFrame({"mz": row["mz_array"], "intensity": row["intensity_array"]})
row_df.plot(
kind="spectrum",
x="mz",
y="intensity",
annotate_mz=True,
bin_method="none",
annotate_top_n_peaks=5,
aggregate_duplicates=True,
title=f"Mass spectrum of {row['sequence']}",
);
We include a residue remapping to ensure our input dataset can be mapped to the format the model vocabulary expects.
model.residue_set.update_remapping(
{
"M(ox)": "M[UNIMOD:35]",
"M(+15.99)": "M[UNIMOD:35]",
"S(p)": "S[UNIMOD:21]", # Phosphorylation
"T(p)": "T[UNIMOD:21]",
"Y(p)": "Y[UNIMOD:21]",
"S(+79.97)": "S[UNIMOD:21]",
"T(+79.97)": "T[UNIMOD:21]",
"Y(+79.97)": "Y[UNIMOD:21]",
"Q(+0.98)": "Q[UNIMOD:7]", # Deamidation
"N(+0.98)": "N[UNIMOD:7]",
"Q(+.98)": "Q[UNIMOD:7]",
"N(+.98)": "N[UNIMOD:7]",
"C(+57.02)": "C[UNIMOD:4]", # Carboxyamidomethylation
"(+42.01)": "[UNIMOD:1]", # Acetylation
"(+43.01)": "[UNIMOD:5]", # Carbamylation
"(-17.03)": "[UNIMOD:385]",
}
)
from instanovo.transformer.data import TransformerDataProcessor
# HuggingFace dataset
ds = sdf.to_dataset(in_memory=True)
processor = TransformerDataProcessor(
model.residue_set,
reverse_peptide=False,
return_str=True,
)
ds = processor.process_dataset(ds)
from torch.utils.data import DataLoader
# When using SpectrumDataFrame, workers and shuffle is handled internally.
dl = DataLoader(ds, batch_size=64, shuffle=False, collate_fn=processor.collate_fn)
batch = next(iter(dl))
batch = {k: v.to(device) if isinstance(v, torch.Tensor) else v for k, v in batch.items()}
peptides = batch["peptides"]
batch.keys()
Decoding¶
We have three options for decoding:
- Greedy Search
- Beam Search
- Knapsack Beam Search
For the best results and highest peptide recall, use Knapsack Beam Search. For fastest results (over 10x speedup), use Greedy Search.
We generally use a beam size of 5 for Beam Search and Knapsack Beam Search, a higher beam size should increase recall at the cost of performance and vice versa.
Note: in our findings, greedy search has similar performance as knapsack beam search at 5% FDR. I.e. if you plan to filter at 5% FDR anyway, use greedy search for optimal performance.
Greedy Search and Beam Search¶
Greedy search is used when num_beams=1, and beam search is used when num_beams>1
from instanovo.inference import BeamSearchDecoder, GreedyDecoder
num_beams = 5 # Change this, defaults are 1 or 5
if num_beams > 1:
decoder = BeamSearchDecoder(model=model)
else:
decoder = GreedyDecoder(model=model)
Knapsack Beam Search¶
Setup knapsack beam search decoder. This may take a few minutes.
from pathlib import Path
from instanovo.constants import MASS_SCALE
from instanovo.inference.knapsack import Knapsack
from instanovo.inference.knapsack_beam_search import KnapsackBeamSearchDecoder
def _setup_knapsack(model: InstaNovo, max_isotope: int = 2) -> Knapsack:
residue_masses = dict(model.residue_set.residue_masses.copy())
for special_residue in list(model.residue_set.residue_to_index.keys())[:3]:
residue_masses[special_residue] = 0
residue_indices = model.residue_set.residue_to_index
return Knapsack.construct_knapsack(
residue_masses=residue_masses,
residue_indices=residue_indices,
max_mass=4000.0,
mass_scale=MASS_SCALE,
max_isotope=max_isotope,
)
knapsack_path = Path("./checkpoints/knapsack/")
if not knapsack_path.exists():
print("Knapsack path missing or not specified, generating...")
knapsack = _setup_knapsack(model)
decoder = KnapsackBeamSearchDecoder(model, knapsack)
print(f"Saving knapsack to {knapsack_path}")
knapsack_path.parent.mkdir(parents=True, exist_ok=True)
knapsack.save(knapsack_path)
else:
print("Knapsack path found. Loading...")
decoder = KnapsackBeamSearchDecoder.from_file(model=model, path=knapsack_path)
Inference time 🚀¶
Evaluating a single batch...
with torch.no_grad():
output = decoder.decode(
spectra=batch["spectra"],
precursors=batch["precursors"],
beam_size=num_beams,
max_length=config["max_length"],
)
preds = output["predictions"]
probs = output["prediction_log_probability"]
Confidence probabilities¶
The model returns per-residue confidences in the form of token log-probabilities. We can visualize these or use them as part of a workflow.
from typing import Optional, Any
import matplotlib.pyplot as plt
import seaborn as sns
import numpy as np
def plot_residue_confidence(sequence: list[str], log_prob: float, token_log_probs: list[float], peptide: Optional[str] = None) -> None:
if not sequence:
return
ticks = list(range(len(sequence)))
token_probabilities = np.exp(token_log_probs[: len(ticks)])
sequence_confidence = np.exp(log_prob)
_, ax = plt.subplots()
bars = sns.barplot(x=ticks, y=token_probabilities, errorbar=None, ax=ax)
# Increase Y-axis limit to create space for text labels
ax.set_ylim(0, max(token_probabilities) * 1.2)
# Add numbers above bars with a slanted angle
for bar, prob in zip(bars.patches, token_probabilities):
height = bar.get_height()
ax.text(
bar.get_x() + bar.get_width() / 2,
float(height) + 0.02,
f"{float(prob):.4f}",
ha="center",
va="bottom",
fontsize=9,
color="black",
rotation=45,
)
# Check if any residue contains a PTM (e.g., "S(+79.97)")
has_ptm = any("(" in res and ")" in res for res in sequence)
# Set labels
x_label = f" Prediction: {''.join(sequence)}"
if peptide is not None:
x_label += f"\nGround truth: {peptide}"
ax.set_xlabel(x_label)
ax.set_ylabel("Confidence Probability")
# Set title with sequence confidence
ax.set_title(
f"Residue Confidence per Position\nSequence Probability: {sequence_confidence:.4f}"
)
# Set X-ticks
ax.set_xticks(ticks)
ax.set_xticklabels(
sequence,
rotation=45 if has_ptm else 0,
ha="right" if has_ptm else "center",
)
plt.show()
For a spectrum that is sequenced correctly, the sequence probability and per-residue probabilities are uniformly high:
plot_residue_confidence(
output["predictions"][-1],
output["prediction_log_probability"][-1],
output["prediction_token_log_probabilities"][-1],
peptides[-1]
)
For another spectrum which is sequenced incorrectly, the sequence probability is low and the per-residue probabilities of the incorrectly sequenced residues (up to isomerism) are lower than of those correctly sequenced:
plot_residue_confidence(
output["predictions"][2],
output["prediction_log_probability"][2],
output["prediction_token_log_probabilities"][2],
peptides[2]
)
These examples suggest the model is fairly well calibrated.
Evaluation¶
from instanovo.utils.metrics import Metrics
metrics = Metrics(model.residue_set, config["isotope_error_range"])
aa_precision, aa_recall, peptide_recall, peptide_precision = metrics.compute_precision_recall(
peptides, preds
)
peptide_recall
Evaluating on the yeast test fold of the nine-species dataset:
from tqdm.notebook import tqdm
preds = []
targs = []
probs = []
for _, batch in tqdm(enumerate(dl), total=len(dl)):
batch = {k: v.to(device) if isinstance(v, torch.Tensor) else v for k, v in batch.items()}
with torch.no_grad():
output = decoder.decode(
spectra=batch["spectra"],
precursors=batch["precursors"],
beam_size=num_beams,
max_length=config["max_length"],
)
preds.extend(output["predictions"])
probs.extend(output["prediction_log_probability"])
targs += list(batch["peptides"])
Evaluation metrics¶
Model performance without filtering:
aa_precision, aa_recall, peptide_recall, peptide_precision = metrics.compute_precision_recall(
targs, preds
)
aa_error_rate = metrics.compute_aa_er(targs, preds)
auc = metrics.calc_auc(targs, preds, np.exp(pd.Series(probs)))
print(f"amino acid error rate: {aa_error_rate:.5f}")
print(f"amino acid precision: {aa_precision:.5f}")
print(f"amino acid recall: {aa_recall:.5f}")
print(f"peptide precision: {peptide_precision:.5f}")
print(f"peptide recall: {peptide_recall:.5f}")
print(f"area under the PR curve: {auc:.5f}")
We can find a threshold to ensure a desired FDR:¶
Model performance at 5% FDR:
fdr = 5 / 100 # Desired FDR
_, threshold = metrics.find_recall_at_fdr(targs, preds, np.exp(probs), fdr=fdr)
aa_precision_fdr, aa_recall_fdr, peptide_recall_fdr, peptide_precision_fdr = (
metrics.compute_precision_recall(targs, preds, np.exp(probs), threshold=threshold)
)
print(f"Performance at {fdr * 100:.1f}% FDR:\n")
print(f"amino acid precision: {aa_precision_fdr:.5f}")
print(f"amino acid recall: {aa_recall_fdr:.5f}")
print(f"peptide precision: {peptide_precision_fdr:.5f}")
print(f"peptide recall: {peptide_recall_fdr:.5f}")
print(f"area under the PR curve: {auc:.5f}")
print(f"confidence threshold: {threshold:.5f} <-- Use this as a confidence cutoff")
Note: to reproduce the results of the paper, the entire Yeast test set should be evaluated with the 0.1.7 release of InstaNovo.
Saving the predictions...¶
pred_df = pd.DataFrame(
{
"targets": targs,
"tokenized_predictions": preds,
"predictions": ["".join(x) for x in preds],
"log_probabilities": probs,
}
)
pred_df.head()
pred_df.to_csv("predictions_knapsack_beam_search.csv", index=False)
InstaNovo+: Iterative Refinement with a Diffusion Model¶
In this section, we show how to refine the predictions from the transformer model with a diffusion model.
First, we download the model checkpoint.
from instanovo.diffusion.multinomial_diffusion import InstaNovoPlus
InstaNovoPlus.get_pretrained()
diffusion_model, diffusion_config = InstaNovoPlus.from_pretrained("instanovoplus-v1.1.0")
diffusion_model = diffusion_model.to(device).eval()
Next we create a decoder object.
from instanovo.inference.diffusion import DiffusionDecoder
diffusion_decoder = DiffusionDecoder(model=diffusion_model)
Then we prepare the inference data loader using predictions from the InstaNovo transformer model.
from instanovo.constants import REFINEMENT_COLUMN
from instanovo.diffusion.data import DiffusionDataProcessor
diffusion_ds = sdf.to_dataset(in_memory=True)
# Add the "tokenized_predictions" column from pred_df to the diffusion_ds HuggingFace dataset
diffusion_ds = diffusion_ds.add_column(REFINEMENT_COLUMN, pred_df["tokenized_predictions"])
diffusion_processor = DiffusionDataProcessor(
diffusion_model.residue_set,
reverse_peptide=False,
return_str=True,
metadata_columns=[REFINEMENT_COLUMN],
)
diffusion_ds = diffusion_processor.process_dataset(diffusion_ds)
diffusion_dl = DataLoader(
diffusion_ds,
batch_size=64,
shuffle=False,
collate_fn=diffusion_processor.collate_fn,
)
Finally, we predict sequences by iterating over the spectra and refining the InstaNovo predictions.
predictions = []
log_probs = []
for batch in tqdm(diffusion_dl, total=len(diffusion_dl)):
batch = {k: v.to(device) if isinstance(v, torch.Tensor) else v for k, v in batch.items()}
with torch.no_grad():
output = diffusion_decoder.decode(
spectra=batch["spectra"],
spectra_padding_mask=batch["spectra_mask"],
precursors=batch["precursors"],
initial_sequence=batch[REFINEMENT_COLUMN],
)
predictions.extend(output["predictions"])
log_probs.extend(output["prediction_log_probability"])
Iterative refinement improves performance on this sample of the Nine Species dataset. (To replicate the performance reported in the paper, you would need to evaluate on the entire dataset.)
(
aa_precision_refined,
aa_recall_refined,
peptide_recall_refined,
peptide_precision_refined,
) = metrics.compute_precision_recall(targs, predictions=predictions)
aa_error_rate_refined = metrics.compute_aa_er(targs, predictions)
auc_refined = metrics.calc_auc(targs, predictions, np.exp(pd.Series(log_probs)))
print(f"amino acid error rate: {aa_error_rate_refined:.5f}")
print(f"amino acid precision: {aa_precision_refined:.5f}")
print(f"amino acid recall: {aa_recall_refined:.5f}")
print(f"peptide precision: {peptide_precision_refined:.5f}")
print(f"peptide recall: {peptide_recall_refined:.5f}")
print(f"area under the ROC curve: {auc_refined:.5f}")
print(f"Decrease in AA error rate: {100 * (aa_error_rate - aa_error_rate_refined):.2f}%")
print(f"Increase in AA precision: {100 * (aa_precision_refined - aa_precision):.2f}%")
print(f"Increase in AA recall: {100 * (aa_recall_refined - aa_recall):.2f}%")
print(f"Increase in peptide precision:{100 * (peptide_precision_refined - peptide_precision):.2f}%")
print(f"Increase in peptide recall: {100 * (peptide_recall_refined - peptide_recall):.2f}%")
print(f"Increase in AUC: {100 * (auc_refined - auc):.2f}%")
diffusion_predictions = pd.DataFrame(
{
"targets": targs,
"tokenized_predictions": predictions,
"predictions": ["".join(x) for x in predictions],
"log_probabilities": log_probs,
}
)
diffusion_predictions.head()
diffusion_predictions.to_csv("diffusion_predictions.csv", index=False)